BGSA
BGSA is a bit-parallel global sequence alignment toolkit for multi-core and many-core architectures. BGSA supports both Myers algorithm (including the banded version) and BitPAl in order to gain fast speed of calculating unit-cost global alignments as well as global alignments with general integer scoring schemes.
Contents
Features
- Supports both unit-cost global aligment(Myers algorithm) and general integer scoring global aligment(BitPAl algorithm).
- Supports both global and semi-global aligment.
- It also implements the banded Myers algorithm for fast verification under a given error threshold e;
- Supports multiple SIMD intrinsics: SSE, AVX2, KNC and AVX512. It also supports heterogeneous platform that contains KNC and CPU.
- The parallel framework can extend to other similar algorithm easily with a little change.
- Super faster than other similar softwares.
Usage
- Step 1: Use the generator program to generate the kernel source with your specified score and architecture. And you need the Java runtime 1.7 or higher.
cd generator java -jar generator.jar -M 2 -I -3 -G -5 -a sse - Step 2: Move the generated file to the BGSA source folder accroding to your selected architecture, and then run
makecommand.mv generated/align_core.c ../original/BGSA_SSE/ cd ../original/BGSA_SSE/ makeThe default compiler is icc and CPU platform. You can pass
arch=KNLfor KNL platform andcc=gccfor other compilers.# use gcc compiler on KNL platform make arch=KNL cc=gcc - Step 3: Run a test.
./aligner -q sample-data/query.txt -d sample-data/subject.txt -f data/result.txt - Step 4: Convert the result file to readable format.
./convert -r data/result.txt
Generator
We provide a code generator written in Java to quickly generate algorithm kernel source for a wide variety of devices as well as different scoring schemes. The following are the parameters of the generator module:
-M,--match <arg>: Specify the match score which should be positive or zero. Default is 2.
-I,--mismatch <arg>: Specify the mismatch score which should be negative. Default is -3.
-G,--gap <arg>: Specify the gap score which should be negative. Default is -5.
-d,--directory <arg>: Specify the directory where to place generated source files.
-n,--name <arg>: Set the name of generated source file.
-t,--type <arg>: Set BitPAl algorithm type. Valid values are: non-packed, packed. Default is packed.
-m,--myers: Using Myers' algorithm. Valid values are: 0, 1. 0 presents weights(0, -1, -1) and 1 presents weights(0, 1, 1). Default is 0.
-a,--arch: Specify the SIMD architecture. Valid values are: none, sse, avx2, knc, avx512. Default is sse. If you want generate kernel source for multiple architectures, you can use '-' to join them as none-sse-avx2
-e,--element: Specify vector element size. Valid value is 64, 32, 16, 8. Default is 32.
-s,--semi: Using semi-global algorithm.
-b,--banded: Using banded myers algorithm.
-h,--help: Display help Information.
Performance
The following figures show the performance comparison of BGSA, Parasail and SeqAn on CPU and Xeon Phi platforms for unit scoring scheme. From the figures, we can see that the performance of BGSA is much faster than that of the other two algorithms. For more performance evluations, you can see our paper(under submission). And you can download test data from this link.
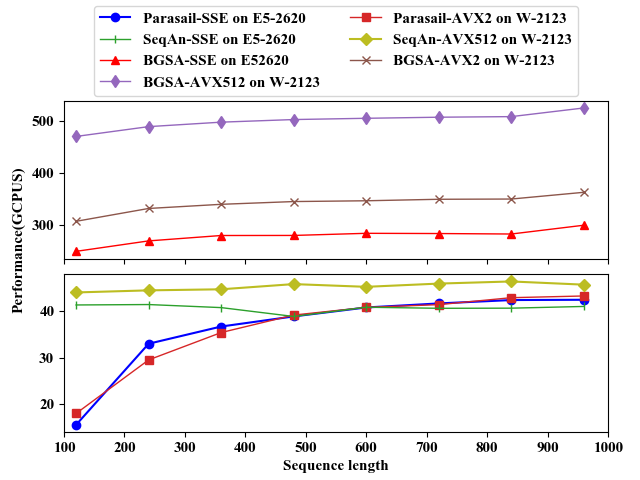
Comparison on E5-2620 and W-2123
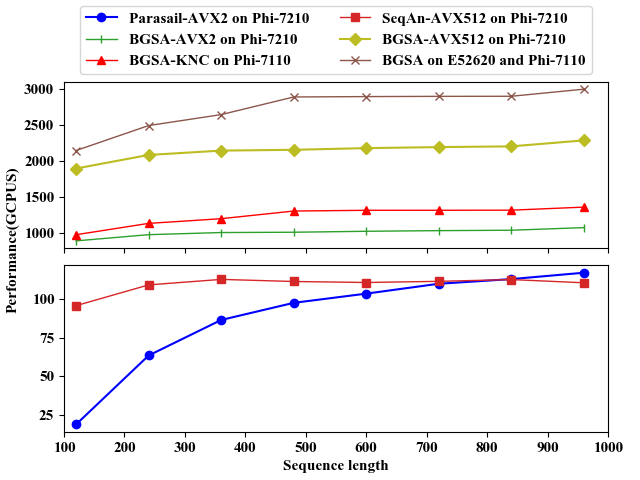
Comparison on Phi-7110 and Phi-7210
Use BGSA in your project
You can use BGSA in your project by directly copying header and source files. For simplicity, you can first save the seqeunces to be compared into temporary files, and then call BGSA to read sequences from the temporary files, calcuate alignment scores and save them to a result file, and finally your program can read the scores from the result file. If you want to use sequences already stored in an array, you can modify the logic in calling get_read_from_file and get_ref_from_file and change the pointer to point the existing sequence array.
If you just want to use the kernel alignment method, you need to process your data into the format required by this method. The following is a complete demo using SSE intrinsics:
demo.c
#include <stdio.h>
#include <string.h>
#include "align_core.h"
int cpu_threads = 1;
void align(char *query, char **subjects, int subject_count) {
int query_len = strlen(query);
int subject_len = strlen(subjects[0]);
int real_subject_count = subject_count;
int total_subject_count = real_subject_count;
if (real_subject_count % SSE_V_NUM != 0) {
total_subject_count = (real_subject_count / SSE_V_NUM + 1) * SSE_V_NUM;
}
seq_t subject_seq;
subject_seq.content = malloc_mem(sizeof(char) * total_subject_count * (subject_len + 1));
subject_seq.count = real_subject_count;
subject_seq.len = subject_len;
for (int i = 0; i < total_subject_count; i++) {
for (int j = 0; j < subject_len + 1; j++) {
subject_seq.content[i * (subject_len + 1) + j] = subjects[i][j];
}
}
int word_num;
if (full_bits) {
word_num = (subject_len + SSE_WORD_SIZE) / (SSE_WORD_SIZE - 1);
} else {
word_num = (subject_len + SSE_WORD_SIZE - 1) / (SSE_WORD_SIZE - 2);
}
int pre_process_size = sizeof(sse_read_t) * word_num * CHAR_NUM * real_subject_count;
sse_read_t *pre_precess_subjects = malloc_mem(pre_process_size);
init_mapping_table();
sse_handle_reads(&subject_seq, pre_precess_subjects, word_num, 0, total_subject_count);
char *pre_precess_query = malloc_mem(sizeof(char) * query_len);
for (int i = 0; i < query_len; i++) {
pre_precess_query[i] = mapping_table[query[i]];
}
sse_write_t *results = malloc_mem(sizeof(sse_write_t) * total_subject_count);
__m128i *bit_mem = malloc_mem(sizeof(__m128i) * word_num * dvdh_len);
align_sse(pre_precess_query, pre_precess_subjects, query_len, subject_len, word_num, 1, 0, results, bit_mem);
for (int i = 0; i < real_subject_count; i++) {
printf("%d\n", results[i]);
}
free_mem(subject_seq.content);
free_mem(pre_precess_subjects);
free_mem(pre_precess_query);
free_mem(results);
free_mem(bit_mem);
}
int main() {
char *query = "AAAA";
char *subjects[4] = {"AAAA", "AACA", "CAAC", "AGGG"};
align(query, subjects, 4);
return 0;
}
You can use the following command to compile demo.c .
icc -o demo demo.c global.c align_core.c -qopenmp
Support or Contact
If you have any questions, please contact: Weiguo,Liu ( weiguo.liu@sdu.edu.cn).